
Global MS Leaders Declare MS is a Preventable Disease to Mark World MS Day
Leading MS organisations, including MS Australia, sign the Nice Declaration, representing an unprecedented commitment to make MS prevention a global priority.

Leading MS organisations, including MS Australia, sign the Nice Declaration, representing an unprecedented commitment to make MS prevention a global priority.

MS Australia and MS Nurses Australasia have today launched ‘Back on Track,’ an innovative new MS Nurse-led video and audio resource designed to empower and guide people living with Multiple Sclerosis (MS) through their unique journey.

You may experience nicotine withdrawal symptoms when you attempt to quit smoking. This affects around 50% of all smokers and occurs regardless of how much you have smoked in the past.

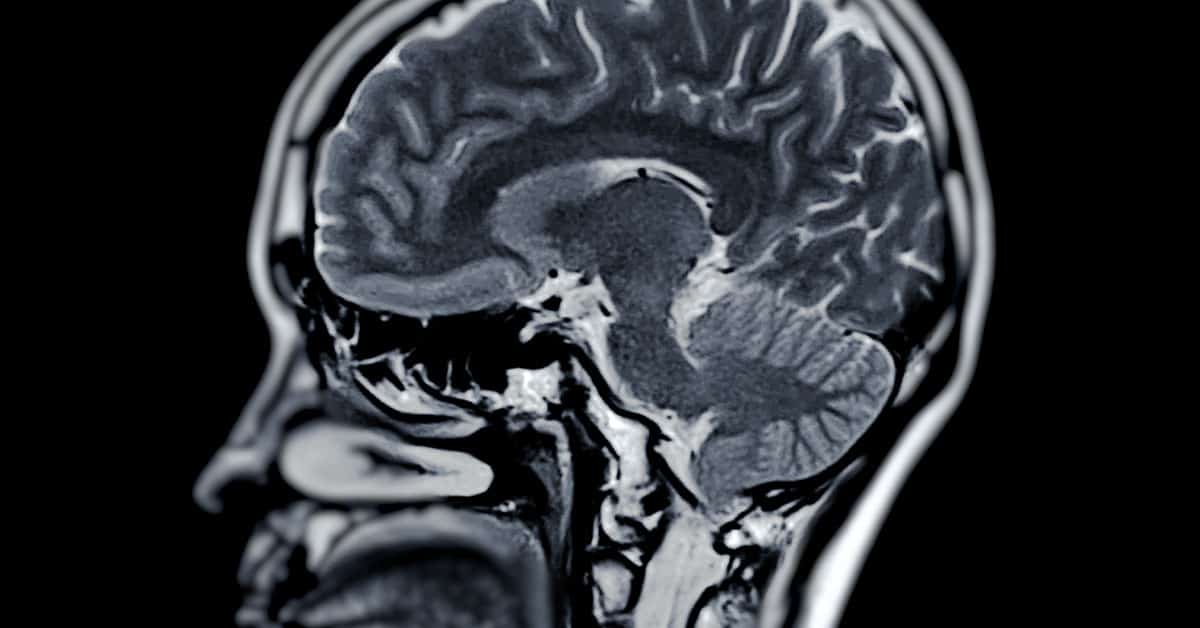
MS and smoking cigarettes both damage the central nervous system, which includes the brain and spinal cord. This damage can be noticed in many different ways, such as sensory disturbances or vision problems.

Studies have shown that smoking can reduce the effectiveness of MS medications. While the exact mechanisms are not yet fully understood, it is considered likely that inhalation of cigarette smoke plays an important role and promotes the formation of antibodies.

CEO Rohan Greenland sends a huge thank you to everyone supporting this year’s May 50K and teases MS Australia’s plans for World MS Day, including the launch of an MS Nurse-led resource and a joint MS prevention statement with MS Canada.

Alex is a young professional from Brisbane living with MS. Diagnosed in 2021 at 25 years old, he reflects on how his diagnosis changed his life and the work he has done to thrive personally and professionally in the years since.

New Australian research shows that people living with MS are more likely to stay in the workforce when they feel safe disclosing their MS at work and are confident in performing their jobs.
People living with progressive MS ensure International Progressive MS Alliance efforts are focused on solutions to result in life-changing treatments.